
Downloads
NAMD performance graphs
Graphs of NAMD performance. The explicit-solvent system has: 47,481 atoms, PME electrostatics, and a 4 fs timestep. These simulations were performed on a 12-core workstation with Xeon E5-2680 CPUs and three GeForce GTX 960 graphics cards. Simulations were performed with NAMD 2.12 (pre-compiled Linux-x86_64-multicore-CUDA or Linux-x86_64-multicore builds) unless otherwise noted.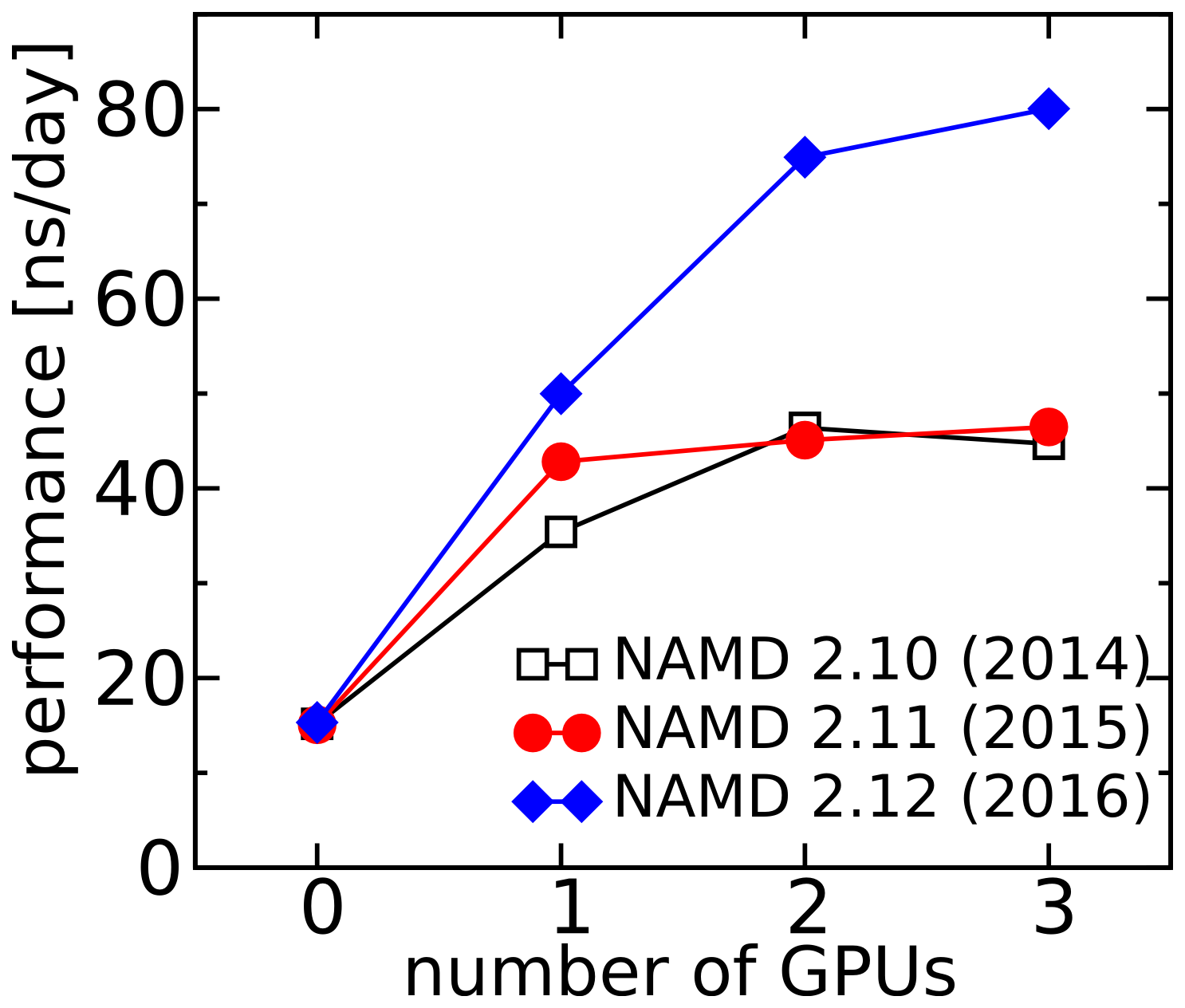
Comparison of recent NAMD versions for 47K-atom system running on 12 CPU cores.
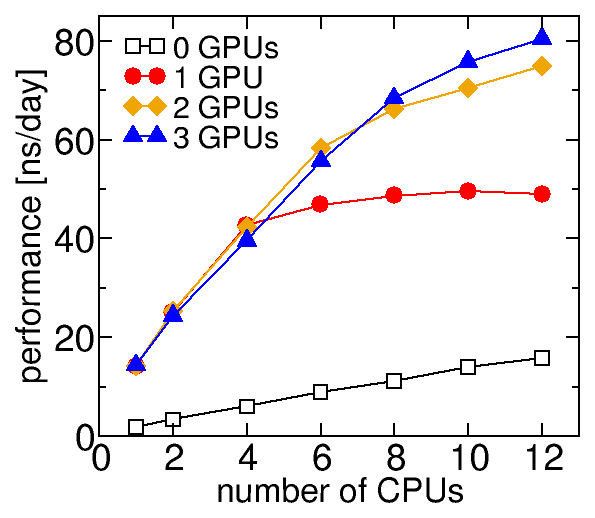
Performance of NAMD 2.12 on with different numbers of cores and GPUs for a 47K-atom system. The cores not being used for the simulation are otherwise unoccupied. There is a cost to occupying the cores. Notably, the highest aggregate performance I can get from this workstation is by running three simulations simultaneously (4 CPUs and 1 GPU eac), which is 100 ns/day. The performance of one of the three simulations (33 ns/day) is less than running a simulation with 4 CPUs and 1 GPU on an otherwise empty node (43 ns/day).
Complete set of files for calculating Ramachandran free-energy landscape of alanine dipeptide on graphene.
Ready-to-run NAMD configuration file: PhiPsi/namd/abf_graph_alad_36m.0.namdDownload files: PhiPsi.zip
Complete set of files for calculating free-energy of adsorption of m-cresol to hydroxylated graphene.
Ready-to-run NAMD configuration file: GraphOhOh/namd/abfFreeTop_3_15_ohoh_bPh.0.namdDownload files: GraphOhOh.zip
DiffusionFusion: a program for calculating position-dependent diffusivity from molecular dynamics trajectories
This code, which works for both conventional and fractional-order Smoluchowski models is available on our GitHub page.The approach is described in DOI: 10.1038/srep35913 and 10.1021/ct300867e
Using Martini polarizable water with NAMD
PSF File: cg_polar_water.psfPDB File: cg_polar_water.pdb
CHARMM-style parameter file: martini_pol_water.par
Example NAMD config file: namd_waterP1.namd
Script for building the PSF and PDB above: makeMartiniPolarWater.tcl
Necessary NAMD modifications:
Change the following line in Molecule.C
} else if (atoms[atom_number-1].mass <=3.5) {} else if (atoms[atom_number-1].mass <=3.5 || strcmp(atom_type,"D")==0 ) {
Comment out this splitPatch line SimParameters.C
else if (!strcasecmp(s, "1-2"))
{
exclude = ONETWO;
//splitPatch = SPLIT_PATCH_POSITION;
}
Comment out this line in SimParameters.C:
//NAMD_die("Do not use Particle Mesh Ewald with Martini. Set: PME off");